CELL2007 Molecular Exploration Project
Group 7

Tertiary Structure Prediction
Figure 9: Predicted Tertiary Structure of TESK1 by PHYRE2. Rendered using PyMol.
[2] Protein structure prediction on the web: a case study using the Phyre server, Kelley LA and Sternberg MJE. Nature Protocols 4, 363 - 371 (2009)
[5] The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.
PHYRE2
PHYRE (Protein Homology/Analogy Recognition Server) is a web based protein structure prediction tool. It uses protein threading to predict the structure. Protein threading differs from homology modelling as it is designed to predict structures with no deposited homologous structures. Specfically it uses statistics to align the given protein sequence to known sequences, and uses the most probable sequence as the model for the predicted structure.
SWISS-MODEL

From Figure 7 it is possible to see that the amino acids within the predicted structure of TESK1 using the PHYRE server do not all fall within the energetically allowed regions of the Ramanchandran plot. The highlighted ARG 608 residue exemplifies this; as, although it falls ithin the left-handed helix region, there is no left-handed helix. Furthermore, there are many residues which are not glycine falling within the disallowed region of the plot- these residues most likely correspond to the disordered region. This plot displays the unlikelihood of the PHYRE prediction being an accurate prediction of TESK1's structure.
Figure 10: Ramanchandran Plot of the Predicted Structure of TESK1 and Rendered Structure of Highlighted ARG 608 Residue. The ramanchandran plot shows a plot of the combinations of the torsion angles phi and psi with the TESK1 predicted structure. Circles represent all amino acids besides glycine, triangles represent glycine. The ARG 608 is highlighted to denote the improbable combination of the torsion angles within left-handed helix and not allowed regions of the plot. Generated using Maestro.
[6] Schrödinger Release 2015-1: Maestro, version 10.1, Schrödinger, LLC, New York, NY, 2015.
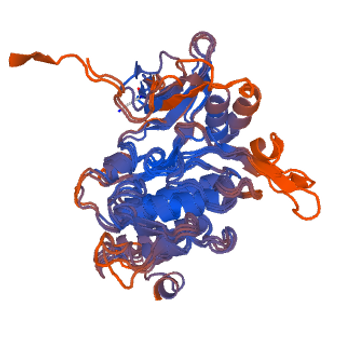
Unlike PHYRE, SWISS-MODEL uses homology modelling to predict protein structures. This is currently the most reliable method of prediction.
The models generated were based upon the LIM Kinase Domain and only covered the region from approximate 50-320 residues of the TESK1 FASTA sequence. This correlates to the above PHYRE prediction, as only the kinase domain structure was successfully modelled. This again illustrates the inefficiency of structure prediction. However, it does show that the TESK1 protein has a kinase domain near its N-terminal region, that perhaps once activated induces a conformational change in its other constituent domains -that have yet to be identified structurally- thereby affecting its function.
Figure 11: A 3D structure of the Kinase Domain of TESK1 predicted using swiss-model, three of the predicted models are superimposed on one another.
[7] Arnold K., Bordoli L., Kopp J., and Schwede T. (2006). The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 22,195-201
3D LIGAND SITE
3D Ligand site uses homologous structures to the query sequence to predict the structure of particular conserved domain, and hence the binding site within that. Specifically, it utilises MAMMOTH to identify homologous structures with a ligand bound, and then single ligand clustering to group the identified ligands.
The predicted structure of TESK1 correlates to the structures above. It again displays the kinase domain, but this time with a possible ligand bound. The binding site is homologous with those of other kinase domains, being situated between the large and small lobes. The areas highlighted in blue indicate residues involved in binding. Within these residues there are conserved amino acids which appear in all kinase domains. For example, there are two Valine residues 71 and 84 which appear in the binding site and could be involved in interacting with the purine ring of the ATP. Furthermore, there are the Lys 86/179 residues which most likely ion pair with the ATP, with the K86 being the essential lysine residue. There are also Glycines residues (64 and 66) which are present in the glycine rich loop. Additionally, there is an Asp 196 residue wihch corresponds to the Asp residue involved in coordinating magnesium in other kinase domains. Using Phospho.ELM the cryptic phosphorylation site was found to be S220 [9]. Finally, the predictions for ligands of TESK1 were concordant with known ligands of kinase domains: peptides, ATP and magnesium.
Figure 12: Predicted Kinase Domain of TESK1 bound to a Possible Ligand. The residues highlighted in blue indicate the predicted residues involved in binding. Created using 3D Ligand Site.
[8] Wass M.N., Kelley L.A. and Sternberg M.J. (2010) 3DLigandSite: predicting ligand-binding sites using similar structures.NAR 38, W469-73.

Only 42% of the amino acid sequence was successfully modelled at >90% confidence for TESK1. This corresponds to the kinase domain.
57% was predicted to be disordered, showing that a complete reliable structure for TESK1 can not be predicted using protein modelling.